Learning from the sequences of the SARS-CoV-2 genome as the virus mutates and spreads
What have we and what can we learn from the sequences of the SARS-CoV-2 genome as the virus mutates and spreads around the world?
May 11, 2020
As background, we would recommend the NYT piece by Jonathan Corum and Carl Zimmer on “How Coronavirus Mutates and Spreads.”
Some of the important things so far from SARS-CoV-2 genome sequences:
Ashley Haase:
Virus Origins
By comparing SARS-CoV-2 sequences with other human coronaviruses, and coronaviruses of animals and bats, SARS-CoV-2 is related to but distinct from SARS-CoV and MERS, the two human coronaviruses that preceded SARS-CoV-2 as causes of acute respiratory illness and severe or fatal pneumonia. SARS-CoV-2’s closest relative (96% identity) is a coronavirus from Rhinophilus affinis bats (RaTG13), but there are critical differences in the SARS-CoV-2 Spike protein that optimize binding to its cellular receptor, ACE-2, that arose through natural selection. While we don’t know if natural selection occurred in an animal host, like the Malayan pangolin in the Wuhan market, or in humans following zoonotic transfer, these critical changes in the SARS-CoV-2 genome make it highly unlikely that SARS-CoV-2 is a laboratory construct or purposefully manipulated virus.
SARS-CoV-2 Mutations and Transmissibility
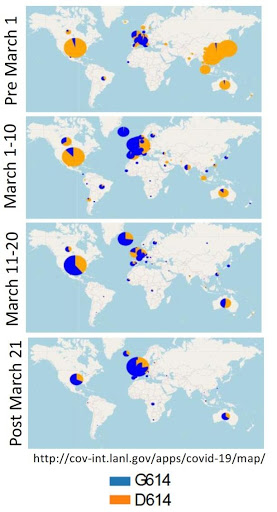
The HIV database team at Los Alamos National Laboratory has developed an analysis pipeline to track the evolution of the SARS-CoV-2 Spike protein, using the Global Initiative for Sharing All Influenza Data (GISAID) SARS-CoV-2 sequence database. They have just published as a preprint: Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2; which Identifies a D614G spike mutant that began spreading in Europe in early February. When introduced into new regions, D614G rapidly becomes the dominant form. The transition from Spike D614 to predominantly G614 in Canada and the USA took place in just a short time from the beginning to the end of March (see map).
The authors suggest that there are two mechanisms that might explain why the D614G mutation is associated with increased transmission: 1) structural changes that facilitate virus entry; and 2) an immunological mechanism called antibody-dependent enhancement whereby antibodies can increase replication in macrophages, as had been shown in SARS-CoV infected rhesus macaques.
SARS-CoV-2 D614G Mutation and Disease Severity
The authors examined clinical outcome data from a single geographic region, Sheffield, England, and compared hospitalization status at the beginning of March, when the D614 form predominated, to the end of March, when G614 predominated. No significant correlation was found in patients requiring hospitalization or admission to the ICU. While D614G did not predict hospitalization, patients with the Spike G614 mutation had significantly higher viral loads.
What are the implications of mutations in the Spike gene for antibody tests? For Vaccine development?
Marc Jenkins: The D614G strain could be an issue for the antibody test, although all 23 hospitalized PCR+ people we tested 10 days after symptom onset made antibodies to the Wuhan S1-RBD. It's likely that SARS-CoV-2 strain sequences would be useful information to have. SARS-CoV-2 could accumulate point mutations that eventually lead to antibody evasion. So, tracking SARS-CoV-2 strain evolution will be critical if it turns out that variants arise that are not neutralized by antibodies to the current virus.
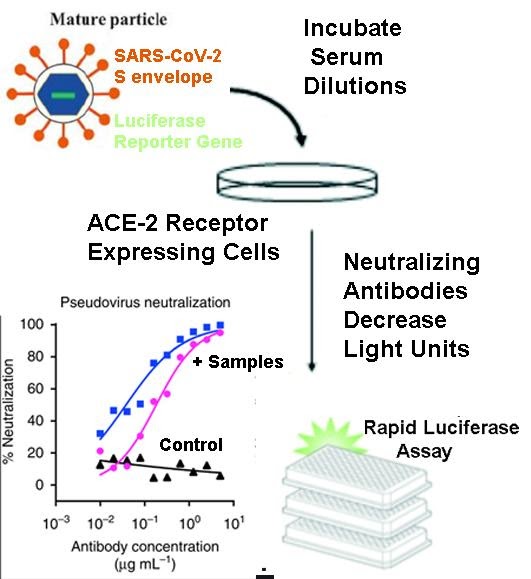
Alon Herschhorn: We are working on pseudotyped virus neutralizing antibody tests that can be done safely under enhanced BSL-2 conditions in MRF. These tests currently mostly use the wild-type SARS-CoV-2 spike to display (pseudotype) the protein on the surface of either HIV-based or VSV-based viral particles. Typically, these viral particles carry a reporter gene that is used to estimate the ability of the virus to infect target cells. The Figure shows a virus pseudotyped with the SARS-CoV-2 Spike (S) protein on its surface, and a luciferase reporter gene that will give off light when target cells bearing the ACE-2 receptor are entered. Neutralizing antibodies that bind to the Spike protein and block that interaction to prevent entry will decrease the amount of light generated as a readout of the assay.
The effect of the D614G change on the conformation of the spike as well as the identity and nature of immunodominant epitopes are still unknown. Conformational changes can alter antibody sensitivity and immunodominant epitopes can lead to dominant responses against specific epitopes. In the SARS-CoV spike, the equivalent D614 residue is embedded in an immunodominant epitope but the ability of this region in SARS-CoV-2 spike to elicit an antibody response is still not known. This information will determine whether vaccine candidates should include both D614 and G614 variants and whether SARS-CoV-2 entry assays using the wild type spike are adequate for understanding the antibody immune response in infected individuals.
How do we test for neutralizing bodies with live virus?
Tyler Bold: Our neutralization assay is similar to Alon’s pseudotyped virus assay in the two step pre-incubation of the virus with dilutions of plasma/serum, followed by a test for in vitro infection activity. However, we are using live virus modified to express Green Fluorescent Protein (GFP) in a high-throughput assay with GFP as a reporter in 96-well plates to rapidly screen many samples. One advantage of using live virus is that S1 RBD is clearly not the only, or even necessarily the most important target of the adaptive immune response to SARS-CoV-2 (Beyond the Spike: identification of viral targets of the antibody response to SARS-CoV-2 in COVID-19 patients). Our live virus assay enables characterization of the functional significance of the broader immune response targeted against multiple viral antigens.
We have the first US strain (USA-WA1/2020), which is D614. We will start using this for in vitro neutralization assays to determine if plasma from convalescent individuals can block the live virus from infecting cells. Once a D614G strain becomes available, we will seek to obtain that as well. Whether or not plasma from individuals infected with D614G strains is still neutralizing against USA-WA1/2020 is an important question that we can test if we have a mechanism for matching a particular serum sample to the viral genotype of the infected patient.
What do we know about SARS-CoV-2 mutations in strains currently circulating in Minnesota?
Daryl Gohl and Kenny Beckman: Thanks to early sequencing efforts by the Minnesota Department of Health, we have insight into the strains circulating in Minnesota and the likely origin of the introductions. Based on an analysis from Nextstrain, a group that aims to provide real-time genomic surveillance of pathogen evolution, there have been multiple introductions of the virus to the state from around the globe, with early introductions coming from either Europe and the East or West Coast of the USA in early March. Based on viral sequencing data from the Minnesota Department of Health and the University of Minnesota, both the D614 and G614 variants are circulating in Minnesota. SARS-CoV-2 strains from Minnesota are highlighted in red in the chart, which presents the viral sequences and strains like a multi-generation family tree.
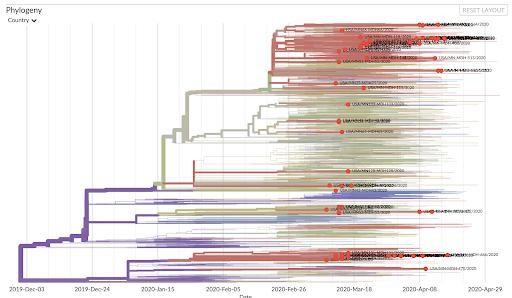
More broadly, what collaborative efforts are underway or planned at the U of MN, Mayo and MDH for sequencing strains and antigenic regions to understand SARS-CoV-2 evolution relevant to patient outcomes, antibody and drug resistance?
Daryl Gohl and Kenny Beckman: The availability of sophisticated genomics tools provides an opportunity to obtain near real-time information about the identity of circulating viral strains and to use this information to inform public health efforts and patient care. We have been working to organize a consortium of scientists from the University of Minnesota, the Mayo Clinic, and the Minnesota Department of Health called “MN-SOS: Minnesota Surveillance of SARS-CoV-2.” This group aims to establish robust and scalable sequencing methods and genomic harmonize data collection efforts across the state, and to sequence SARS-CoV-2 genomes at large scale in order to complement large-scale sequencing and surveillance efforts occurring elsewhere in the world that will enable researchers to link genomic information with clinical or biological features of the virus. The University of Minnesota Genomics Center is also part of the CDC’s SPHERES consortium (SARS-CoV-2 Sequencing for Public Health Emergency Response, Epidemiology, and Surveillance).
Where should these sequencing efforts be headed in the future?
Daryl Gohl & Kenny Beckman: Right now national attention is on PCR and serological testing, and it's fitting that the country is focused on those challenges, including the UMGC. But, I strongly suspect that before long, we'll need to be able to understand outbreaks, differential outcomes, immune responses, and of course vaccine development and refinement by:
- affordable, routine, population-scale deep sequencing of strains
- targeted deep sequencing of antigenic regions
- viral quasispecies analysis including viral burden and intra-host evolution data
In our opinion, sequencing is only the first step. We need high throughput tools to assess the biological function of these changes and it will be very valuable to have a repository of different strains, and available Spike-expressing plasmids of dominant changes. One way that we are now pursuing this direction is by introducing the D614G and other mutations in the CoV-2 spike and testing the effect of these changes on the function and sensitivity to antibody neutralization. Professor Wei-Shou Hu in the College of Science and Engineering is also introducing these mutations in the soluble CoV-2 spike and will construct a stable cell line that expresses these soluble variants. Information on whether these changes confer resistance to antibodies and small molecules will lead to new tests to detect the presence of these alterations. It is not expected that every change will lead to resistance so there is a need to correlate sequence changes with antigenic effects at a large scale. Antigenic “cartography” may be useful to study antigenic differences and distances.
Update by:
Dr. Ashley Haase, MD
Regents’ Professor and Head, Department of Microbiology and Immunology, Professor of Medicine- Infectious Diseases and Internal Medicine
Daryl Gohl, PhD
Group Leader, UMGC Innovation Lab
Ryan Langlois, PhD
Assistant Professor McKnight Presidential Fellow, Department of Microbiology and Immunology
Marc Jenkins, PhD
Regents and Distinguished McKnight University Professor, Department of Microbiology and Immunology
Alon Hershhorn, PhD
Assistant Professor of Medicine, Division of Infectious Diseases and International Medicine
Tyler Bold, MD, PhD
Assistant Professor of Medicine, Division of Infectious Diseases and International Medicine